
Construction and validation of prognostic risk model for patients with hepatocellular carcinoma based on bioinformatics analysis
-
摘要:目的 利用公共数据库构建用于临床治疗肝细胞癌(HCC)的预后风险模型。方法 分别从癌症基因组图谱(TCGA)和国际癌症基因组联盟(ICGC)下载HCC以及癌旁正常组织的mRNA表达数据及临床信息。在TCGA队列中筛选与总生存期(OS)相关的差异表达基因(DEGs), 从中抽取2个或3个mRNAs构成一个组合, 对所有组合进行Cox风险比例回归模型构建。通过受试者工作特征(ROC)曲线的曲线下面积(AUC)确定最优基因组合,并进行基于ICGC队列的外部验证; 以TCGA队列的风险评分中位值将患者分为高风险组与低风险组,进行基因集富集分析(GSEA), 并通过pRRophetic R软件包预测HCC患者使用索拉非尼、丝裂霉素、依托泊苷、阿霉素、紫杉醇和顺铂的相对半抑制浓度(IC50)。结果 该预后风险模型预测TCGA队列的1、3、5年OS的ROC的AUC分别是0.786、0.713、0.699, 预测ICGC队列的1、3、4年OS的ROC的AUC分别为0.719、0.709、0.766。GSEA表明高风险组患者细胞周期相关通路被激活,胆汁酸代谢被抑制。索拉非尼在低风险组的IC50低于高风险组,而细胞周期相关化疗药物在低风险组的IC50高于高风险组,差异均有统计学意义(P < 0.05)。结论 本研究建立并验证了HCC预后风险模型,为HCC患者个体化诊疗方案的制订提供参考依据。Abstract:Objective To construct a prognostic risk model for clinical treatment of hepatocellular carcinoma (HCC) based on public databases.Methods The mRNA expression data and clinical information of HCC and adjacent normal tissues were downloaded from The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC). Differentially expressed genes (DEGs) related to overall survival (OS) were screened in the TCGA cohort, 2 or 3 mRNAs were selected to form a combination, and Cox risk proportional regression model was constructed for all combinations. The optimal gene combination was determined by the area under the curve (AUC) of the receiver operating characteristic (ROC) curve, and external validation based on ICGC cohort was carried out. The patients were divided into high-risk group and low-risk group according to the median risk score of TCGA cohort, gene set enrichment analysis (GSEA) was performed, and the relative half-inhibitory concentrations (IC50) of sorafenib, mitomycin, etoposide, adriamycin, paclitaxel and cisplatin in HCC patients were predicted by pRRophetic R software package.Results For this prognostic risk model, the AUC of the ROC curve for predicting 1-, 3- and 5-year OS in the TCGA cohort were 0.786, 0.713 and 0.699, respectively, and the AUC of the ROC curve for predicting 1-, 3- and 4-year OS in the ICGC cohort were 0.719, 0.709 and 0.766, respectively. GSEA revealed that cell cycle related pathways were activated and bile acid metabolism was inhibited in the high-risk group. The IC50 of sorafenib in the low-risk group was significantly lower than that in the high-risk group, while the IC50 of cell cycle related chemotherapy drugs in the low-risk group was significantly higher than that in the high-risk group (P < 0.05).Conclusion This study establishes and verifies the prognostic risk model for HCC, and provides a reference for the formulation of individualized diagnosis and treatment plan for HCC patients.
-
在全球范围内,肝癌的发病率位居所有新发肿瘤的第6位,是肿瘤相关死亡的重要原因[1]。肝细胞癌(HCC)是最常见的原发性肝癌,常见的病因有慢性乙型肝炎病毒(HBV)或丙型肝炎病毒(HCV)感染、酗酒、非酒精性脂肪性肝病以及黄曲霉毒素中毒等[2]。虽然HCC的治疗取得了一定的进展,但HCC患者的5年存活率仍不足17%[3]。早期HCC患者接受手术治疗或射频消融术后,5年内肿瘤复发率仍高达70%[4]。此外,由于HCC存在显著的异质性,同一分期的HCC患者的预后也可能不同,并且不同患者对同种治疗药物的受益也可能不同[5-7]。正是因为HCC复杂的病因、治疗的局限性及HCC异质性,导致HCC患者预后的预测以及治疗药物的选择更具挑战性,因此有必要寻求更多的可用于指导临床治疗以及预后评估的生物学标志物。本研究基于癌症基因组图谱(TCGA)和国际癌症基因组联盟(ICGC)数据库,确定了与HCC患者预后相关的关键基因,并进行了预后风险模型的构建与验证,现将结果报告如下。
1. 材料与方法
1.1 数据来源
从TCGA(https://portal.gdc.cancer.gov/repository)数据库下载了374例HCC组织及50例癌旁正常组织的转录组数据和对应的临床资料。从ICGC(https://dcc.icgc.org/projects/LIRI-JP) 数据库下载了231例HCC组织的转录组数据和对应的临床资料。TCGA队列中ID为“TCGA-ZS-A9CF”和“TCGA-DD-AACA”的患者分别重复测序3次和2次,因此取“TCGA-ZS-A9CF”和“TCGA-DD-AACA”两类患者多次测序数据的平均值纳入本研究。此外,进一步去除生存资料不完整的样本,最终TCGA队列及ICGC队列分别有365例和231例样本纳入本研究。
1.2 研究方法
在TCGA队列中,首先利用Stats R软件包的wilcox.test以及p.adjust函数确定在癌旁正常组织与肿瘤组织之间的差异表达基因(DEGs)[错误发现率(FDR) < 0.01, log2|fold change| > 2], 然后使用Survival R软件包的coxph函数对DEGs进行基于总生存期(OS)的单因素回归分析(P < 0.05)。利用Combinat R软件包的combn函数生成包含2个与OS相关DEGs或3个与OS相关DEGs的所有基因组合,然后使用Survival R软件包的coxph函数和TimeROC R软件包的timeROC函数批量对所有基因组合进行多因素Cox回归,计算患者的风险评分以及1、3、5年OS的受试者工作特征(ROC)曲线的曲线下面积(AUC)。满足1、3、5年OS的ROC的AUC之和最大的组合为最优组合,对该组合进行基于ICGC队列的外部验证。以TCGA队列风险评分的中位数将TCGA队列及ICGC队列的患者分为高风险组与低风险组,利用Survival R软件包的survdiff函数评估高、低风险组之间的预后差异。使用R语言rms软件包构建列线图并绘制校准曲线以评估列线图的预测性能。利用clusterProfiler R软件包对高、低风险组的基因表达谱进行基因集富集分析(GSEA), 以京都基因和基因组百科全书(KEGG)、特征基因集或效应基因集(HALLMARK)进行注释。最后,通过pRRophetic R软件包预测HCC患者使用索拉非尼、丝裂霉素、依托泊苷、阿霉素、紫杉醇和顺铂的相对半抑制浓度(IC50)[8]。
1.3 统计学分析
使用Wilcoxon检验筛选在肿瘤组织和癌旁组织之间的DEGs, 并采用BH法对P值进行矫正,筛选标准为FDR < 0.01和log2|fold change| > 2。采用χ2卡方检验评估各种临床资料在高、低风险组的分布差异。Kaplan-Meier分析和Log-rank检验评估高、低风险组患者的生存差异。通过单因素和多因素Cox回归分析来确定OS的独立预后因子。所有统计分析均使用R软件(版本4.0.2)。P < 0.05为差异有统计学意义。
2. 结果
2.1 HCC患者的临床病理资料
本研究纳入了TCGA队列及ICGC队列的HCC患者分别为365例、231例,患者临床病理资料见表 1。
表 1 HCC患者的临床病理资料[n(%)]临床资料 TCGA队列
(n=365)ICGC队列
(n=231)年龄/岁 61(16, 90) 69(31, 89) 性别 女 119(32.6) 61(26.4) 男 246(67.4) 170(72.6) 肿瘤分级 Ⅰ级 55(15.1) — Ⅱ级 175(47.9) — Ⅲ级 118(32.3) — Ⅳ级 12(3.3) — 未知 5(1.4) — 肿瘤分期 Ⅰ期 170(46.6) 36(15.6) Ⅱ期 84(23.0) 105(45.5) Ⅲ期 83(22.7) 71(30.7) Ⅳ期 4(1.1) 19(8.2) 未知 24(6.6) 0 血管浸润 有 106(29.0) — 无 205(56.2) — 未知 54(14.8) — 甲胎蛋白 ≤200 ng/mL 201(55.1) — > 200 ng/mL 75(20.5) — 未知 89(24.4) — 年龄以中位数(最小值,最大值)表示。 2.2 最优基因组合分析
经差异分析共确定2 469个DEGs[FDR < 0.01, log2|fold change| > 2]。经单因素Cox回归分析共有5 856个基因与OS相关,最终确定了692个与OS相关的DEGs。随机抽取692个与OS相关的DEGs中的2个,共有239 086种组合,各个组合对应的1、3、5年的AUC均较低。进一步对692个基因随机抽取3个,共有54 989 780种组合,其中最优组合为脑选择性蛋白激酶1(BRSK1)、同源异型盒基因D9(HOXD9)及核转运蛋白α2(KPNA2)。见图 1。
![]() 图 1 最优基因组合分析A: 与OS相关的差异基因的维恩图; B: BRSK1、KPNA2、HOXD9均在HCC组织中高表达;
图 1 最优基因组合分析A: 与OS相关的差异基因的维恩图; B: BRSK1、KPNA2、HOXD9均在HCC组织中高表达;
C: BRSK1、KPNA2、HOXD9与TCGA队列患者OS的单因素Cox分析。2.3 模型普遍适用性的验证
HCC患者风险评分的计算公式为: 风险评分=0.418 57×BRSK1表达水平+0.445 52×KPNA2表达水平+0.293 62×HOXD9表达水平。以TCGA队列风险评分的中位值(0.961)为截断值将TCGA队列的患者分为高风险组与低风险组(图 2A)。TCGA队列中死亡的患者大多集中在高风险组(图 2B)。生存曲线表明高风险组患者较低风险组患者的OS更短,差异有统计学意义(P < 0.05)(图 2C)。该模型预测TCGA队列的1、3、5年OS的ROC曲线的AUC分别是0.786、0.713、0.699(图 2D)。
![]() 图 2 模型预测HCC患者生存情况图A: TCGA队列中风险评分的分布及其中位值; B: TCGA队列中生存状态、生存时间和风险评分的分布; C: TCGA队列中高风险组患者与低风险组患者OS的比较; D: 时间依赖性ROC曲线的AUC评估风险评分预测TCGA队列患者OS的表现; E: ICGC队列中风险评分的分布及其中位值; F: ICGC队列中生存状态、生存时间和风险评分的分布; G: ICGC队列中高风险组患者与低风险组患者OS的比较; H: 时间依赖性ROC曲线的AUC评估风险评分预测ICGC队列患者OS的表现。
图 2 模型预测HCC患者生存情况图A: TCGA队列中风险评分的分布及其中位值; B: TCGA队列中生存状态、生存时间和风险评分的分布; C: TCGA队列中高风险组患者与低风险组患者OS的比较; D: 时间依赖性ROC曲线的AUC评估风险评分预测TCGA队列患者OS的表现; E: ICGC队列中风险评分的分布及其中位值; F: ICGC队列中生存状态、生存时间和风险评分的分布; G: ICGC队列中高风险组患者与低风险组患者OS的比较; H: 时间依赖性ROC曲线的AUC评估风险评分预测ICGC队列患者OS的表现。通过上述计算公式计算ICGC队列各个患者的风险评分,同样以0.961为截断值将ICGC队列的患者分为高风险组与低风险组(图 2E)。与TCGA队列的结果相似, ICGC队列中死亡的患者也同样集中在高风险组(图 2F),生存曲线表明高风险组患者的OS更短,差异有统计学意义(P < 0.05)(图 2G)。鉴于ICGC队列中仅有2例患者OS超过5年,因此仅计算1、3、4年的AUC, 分别为0.719、0.709、0.766(图 2H)。
2.4 风险评分与临床病理资料的相关性
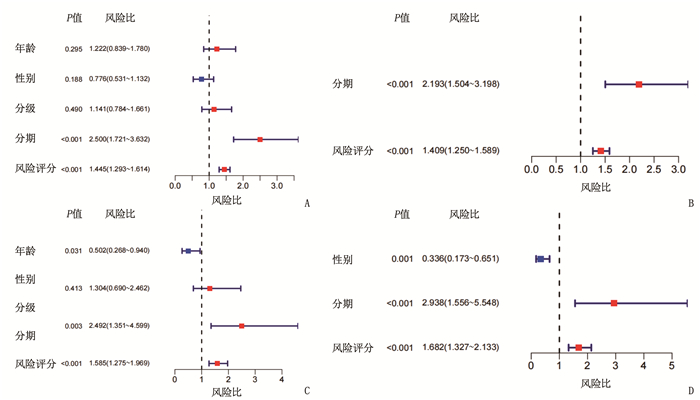
在TCGA队列中,风险评分与患者肿瘤分期、分级、甲胎蛋白及血管浸润具有相关性(P < 0.05); 在ICGC队列中,风险评分与肿瘤分期具有相关性(P < 0.05)。见表 2。在单因素Cox回归分析中, 2个队列的风险评分均与OS相关(TCGA队列: HR=1.445, 95%CI=1.293~1.614, P < 0.001; ICGC队列: HR=1.585, 95%CI=1.275~1.969, P < 0.001)。进一步去除与OS不相关的变量,多因素Cox回归分析表明风险评分在TCGA及ICGC队列中均为OS的独立预后因子(TCGA队列: HR=1.409, 95%CI=1.250~1.589, P < 0.001; ICGC队列: HR=1.682, 95%CI=1.327~2.133, P < 0.001)。见图 3。
表 2 风险评分与HCC患者临床病理资料的相关性临床资料 TCGA队列 ICGC队列 高风险组 低风险组 P 高风险组 低风险组 P 性别 0.80 1.00 女性 58 61 22 39 男性 125 121 62 108 年龄 0.80 0.42 < 60岁 81 84 29 60 ≥60岁 102 98 55 87 肿瘤分级 < 0.05 - 1、2级 134 96 - - 3、4级 47 83 - - 肿瘤分期 < 0.05 < 0.05 Ⅰ、Ⅱ期 141 113 63 78 Ⅲ、Ⅳ期 30 57 21 69 血管浸润 < 0.05 - 无 124 81 - - 有 44 62 - - 甲胎蛋白 < 0.05 - ≤200 ng/mL 122 79 - - >200 ng/mL 27 48 - - ![]() 图 3 风险评分、临床病理资料与OS的相关性分析图A、B: 风险评分、临床病理资料与TCGA队列患者OS的单因素及多因素Cox分析; C、D: 风险评分、临床病理资料与ICGC队列患者OS的单因素及多因素Cox分析。
图 3 风险评分、临床病理资料与OS的相关性分析图A、B: 风险评分、临床病理资料与TCGA队列患者OS的单因素及多因素Cox分析; C、D: 风险评分、临床病理资料与ICGC队列患者OS的单因素及多因素Cox分析。2.5 列线图的构建与验证
本研究基于TCGA队列构建了预测HCC患者OS的列线图,对该列线图进行的内部验证(TCGA队列)及外部验证(ICGC队列)均表明其具有良好的预测性能,见图 4。
![]() 图 4 基于TCGA队列构建的预测HCC患者OS的列线图A: 预测TCGA队列患者OS的列线图; B: 列线图基于TCGA队列的内部验证; C: 列线图基于ICGC队列的外部验证。
图 4 基于TCGA队列构建的预测HCC患者OS的列线图A: 预测TCGA队列患者OS的列线图; B: 列线图基于TCGA队列的内部验证; C: 列线图基于ICGC队列的外部验证。2.6 TCGA和ICGC队列的功能富集分析
基于KEGG及HALLMARK注释的GSEA均表明,与低风险组相比,高风险组细胞周期相关通路被激活(FDR < 0.05), 而胆汁酸代谢在高风险组被抑制(FDR < 0.05), 见图 5。
2.7 TCGA队列高、低风险组患者对化疗及靶向药物的敏感度
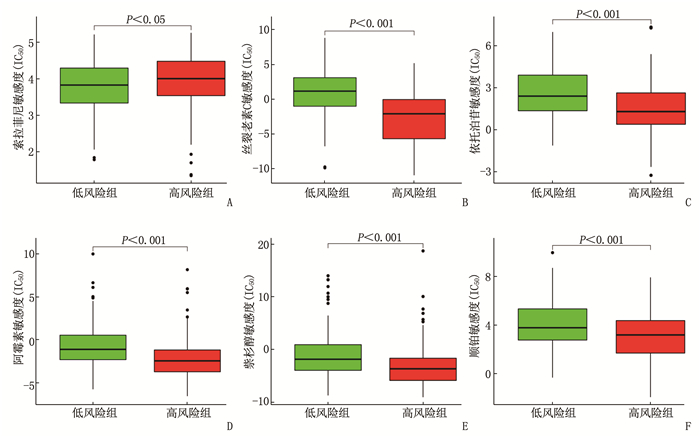
高风险组索拉非尼的相对IC50高于低风险组,而丝裂霉素、依托泊苷、阿霉素、紫杉醇和顺铂的相对IC50低于低风险组,差异均有统计学意义(P < 0.05), 见图 6。
![]() 图 6 TCGA队列高、低风险组患者对化疗及靶向药物的敏感度A: 2组索拉非尼的相对IC50; B: 2组丝裂霉素的相对IC50; C: 2组依托泊苷的相对IC50; D: 2组阿霉素的相对IC50; E: 2组紫杉醇的相对IC50; F: 2组顺铂的相对IC50。
图 6 TCGA队列高、低风险组患者对化疗及靶向药物的敏感度A: 2组索拉非尼的相对IC50; B: 2组丝裂霉素的相对IC50; C: 2组依托泊苷的相对IC50; D: 2组阿霉素的相对IC50; E: 2组紫杉醇的相对IC50; F: 2组顺铂的相对IC50。3. 讨论
随着基因测序技术的飞速发展,许多与HCC预后相关的肿瘤标志物已被发现[9-11], 但是这些标志物的数量仍然有限,因此迫切需要寻找更多肿瘤标志物用于预测HCC患者的预后和指导制订临床个体化的治疗方案。与单基因的标志物相比,基于多个基因的标志物能够更有效地诊断疾病及预测预后[12-13]。目前多数研究[14-17]仅围绕着免疫、代谢、自噬等单方面因素进行多基因的预后预测模型的建立,这就存在着不可避免的缺陷,因为肿瘤的发生发展是多种通路参与的过程。随着机器学习相关领域的发展,诸如Lasso回归、随机森林、支持向量机等一系列算法被开发出来。目前大部分国外相关建模研究[18-20]均运用这些算法对高维数据进行降维,并使用Cox风险比例模型进行建模,但是基于这些算法所建立的模型的变量往往较多,增加了定向测序的成本。此外,国内学者的大部分类似研究[21-22]仅进行了建模后的内部验证,尽管其在内部验证的结果较好,但并未进行外部验证,难以证明所建立的模型具有良好的普遍适用性。
本研究全面评估了基于2个mRNAs、3个mRNAs的所有预后风险模型的预测性能,最终筛选了最优基因组合(BRSK1、HOXD9、KPNA2)。该模型基于TCGA队列的1、3、5年AUC分别是0.786、0.713、0.699, 基于ICGC队列的1、3、4年的AUC分别为0.719、0.709、0.766, 均表明该模型具有良好的预测能力。2个队列的生存曲线表明高风险组患者的OS均低于低风险组患者,提示本模型可以对HCC患者进行有效的危险分层。本研究进一步对TCGA队列高、低风险组患者进行相对IC50预测,表明该模型或可提高治疗的针对性。本研究建立的模型的优势有: ①该模型是基于692个与OS相关的DEGs, 涉及肿瘤多种信号通路; ②该模型仅包括3个mRNAs, 减少了定向测序的成本; ③本研究进行了外部验证,有效证明了本模型具有较好的普遍适用性; ④该模型有助于临床医师制订个体化治疗方案。
HOXD9以及KPNA2已有与HCC的相关报道,而BRSK1与肿瘤的相关报道较少。HOXD9是最靠近染色体3′端的HOXD基因之一,参与前肢和中轴骨骼的发育[23]。本研究表明HOXD9在HCC中高表达,并且与OS相关,与LONG J Y等[24]研究一致。HOXD9过表达可显著促进肝癌细胞的迁移、侵袭和转移,而沉默HOXD9则抑制这些过程,同时HOXD9通过转录因子E盒结合锌指蛋白1(ZEB1)促进HCC上皮-间质过渡和转移[25]。KPNA2是核转运蛋白α家族的成员,在介导核质运输方面起着重要作用[26]。体外和体内实验[27]表明,敲除KPNA2会降低HCC细胞的迁移和增殖能力,同时会过度表达KPNA2而增强HCC的恶性程度。JIANG P等[28]研究发现,在正常肝组织中未观察到KPNA2的表达,因此KPNA2可能作为HCC治疗的潜在靶标。BRSK1是一种一磷酸腺苷(AMP)激活的丝氨酸/苏氨酸激酶,在大脑中特异性表达,在神经元极化中起着重要作用[29]。BRSK1通过促进γ-微管蛋白71磷酸化调控中心体复制,因此BRSK1在调节细胞周期进程中起着重要作用[29]。BRSK1与HCC甚至肿瘤的相关报道较少,本研究证明了BRSK1在HCC中高表达,并且与OS相关,然而其在HCC中的具体生物学作用尚不明确,需要后续研究进一步证实。
为了进一步探讨高风险组HCC患者预后差的潜在机制,本研究对TCGA高、低风险组的基因表达谱数据进行了GSEA。GSEA表明胆汁酸代谢在高风险组被显著抑制,而既往研究[30]证明了胆汁酸在肝内淤积有助于肝癌的发生发展,因此该通路的抑制或是高风险组患者预后差的原因之一。细胞周期相关通路在高风险组激活,例如细胞周期、DNA复制、细胞周期G2/M检验点以及E2F通路,该发现与既往研究[31]是一致的。此外, KRAS信号通路同样在高风险组激活,而既往研究[32]证明KRAS信号通路激活了下游细胞周期通路。细胞周期主要包括DNA合成期(S期)和有丝分裂期(M期),这2个时期被GAP1(G1)和GAP2(G2)期分隔,然而细胞周期调控紊乱被认为是肝癌发生及发展的重要原因之一[33-35], 因此细胞周期相关的化疗药物治疗高风险组患者可能更有效。pRRophetic算法是基于抗癌药物敏感性基因组学数据库(GDSC)中癌细胞系的基因表达谱和药物敏感性数据建立统计模型,并可应用于原发性肿瘤的药物敏感性的预测[8]。本研究利用pRRophetic算法评估了TCGA队列中高、低风险组患者的IC50,发现细胞周期相关的化疗药物如丝裂霉素、依托泊苷、阿霉素、紫杉醇和顺铂等在高风险组患者的相对IC50均低于低风险组,进一步证实了高风险组患者使用细胞周期相关药物的收益可能更高。
综上所述,本研究基于生物信息学方法构建了预后风险模型,该模型可用于评估HCC患者的预后以及相关药物的治疗收益,从而有助于临床医师制订个性化的治疗方案,提高HCC患者生存率。
-
![]()
图 1 最优基因组合分析
A: 与OS相关的差异基因的维恩图; B: BRSK1、KPNA2、HOXD9均在HCC组织中高表达;
C: BRSK1、KPNA2、HOXD9与TCGA队列患者OS的单因素Cox分析。![]()
图 2 模型预测HCC患者生存情况图
A: TCGA队列中风险评分的分布及其中位值; B: TCGA队列中生存状态、生存时间和风险评分的分布; C: TCGA队列中高风险组患者与低风险组患者OS的比较; D: 时间依赖性ROC曲线的AUC评估风险评分预测TCGA队列患者OS的表现; E: ICGC队列中风险评分的分布及其中位值; F: ICGC队列中生存状态、生存时间和风险评分的分布; G: ICGC队列中高风险组患者与低风险组患者OS的比较; H: 时间依赖性ROC曲线的AUC评估风险评分预测ICGC队列患者OS的表现。
![]()
图 3 风险评分、临床病理资料与OS的相关性分析图
A、B: 风险评分、临床病理资料与TCGA队列患者OS的单因素及多因素Cox分析; C、D: 风险评分、临床病理资料与ICGC队列患者OS的单因素及多因素Cox分析。
![]()
图 4 基于TCGA队列构建的预测HCC患者OS的列线图
A: 预测TCGA队列患者OS的列线图; B: 列线图基于TCGA队列的内部验证; C: 列线图基于ICGC队列的外部验证。
![]()
图 6 TCGA队列高、低风险组患者对化疗及靶向药物的敏感度
A: 2组索拉非尼的相对IC50; B: 2组丝裂霉素的相对IC50; C: 2组依托泊苷的相对IC50; D: 2组阿霉素的相对IC50; E: 2组紫杉醇的相对IC50; F: 2组顺铂的相对IC50。
表 1 HCC患者的临床病理资料[n(%)]
临床资料 TCGA队列
(n=365)ICGC队列
(n=231)年龄/岁 61(16, 90) 69(31, 89) 性别 女 119(32.6) 61(26.4) 男 246(67.4) 170(72.6) 肿瘤分级 Ⅰ级 55(15.1) — Ⅱ级 175(47.9) — Ⅲ级 118(32.3) — Ⅳ级 12(3.3) — 未知 5(1.4) — 肿瘤分期 Ⅰ期 170(46.6) 36(15.6) Ⅱ期 84(23.0) 105(45.5) Ⅲ期 83(22.7) 71(30.7) Ⅳ期 4(1.1) 19(8.2) 未知 24(6.6) 0 血管浸润 有 106(29.0) — 无 205(56.2) — 未知 54(14.8) — 甲胎蛋白 ≤200 ng/mL 201(55.1) — > 200 ng/mL 75(20.5) — 未知 89(24.4) — 年龄以中位数(最小值,最大值)表示。  下载: 导出CSV
下载: 导出CSV
表 2 风险评分与HCC患者临床病理资料的相关性
临床资料 TCGA队列 ICGC队列 高风险组 低风险组 P 高风险组 低风险组 P 性别 0.80 1.00 女性 58 61 22 39 男性 125 121 62 108 年龄 0.80 0.42 < 60岁 81 84 29 60 ≥60岁 102 98 55 87 肿瘤分级 < 0.05 - 1、2级 134 96 - - 3、4级 47 83 - - 肿瘤分期 < 0.05 < 0.05 Ⅰ、Ⅱ期 141 113 63 78 Ⅲ、Ⅳ期 30 57 21 69 血管浸润 < 0.05 - 无 124 81 - - 有 44 62 - - 甲胎蛋白 < 0.05 - ≤200 ng/mL 122 79 - - >200 ng/mL 27 48 - -
下载: 导出CSV
-
[1] VILLANUEVA A. Hepatocellular carcinoma[J]. N Engl J Med, 2019, 380(15): 1450-1462. doi: 10.1056/NEJMra1713263
[2] YANG J D, HAINAUT P, GORES G J, et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management[J]. Nat Rev Gastroenterol Hepatol, 2019, 16(10): 589-604. doi: 10.1038/s41575-019-0186-y
[3] TORRE L A, BRAY F, SIEGEL R L, et al. Global cancer statistics, 2012[J]. CA Cancer J Clin, 2015, 65(2): 87-108. doi: 10.3322/caac.21262
[4] BROWN Z J, GRETEN T F, HEINRICH B. Adjuvant treatment of hepatocellular carcinoma: prospect of immunotherapy[J]. Hepatology, 2019, 70(4): 1437-1442. doi: 10.1002/hep.30633
[5] HUANG W T, SKANDERUP A J, LEE C G. Advances in genomic hepatocellular carcinoma research[J]. Gigascience, 2018, 7(11): 1-22. http://www.onacademic.com/detail/journal_1000041617495499_6d02.html
[6] DOMINGUEZ D A, WANG X W. Impact of next-generation sequencing on outcomes in hepatocellular carcinoma: how precise are we really[J]. J Hepatocell Carcinoma, 2020, 7: 33-37. doi: 10.2147/JHC.S217948
[7] CARUSO S, O'BRIEN D R, CLEARY S P, et al. Genetics of hepatocellular carcinoma: approaches to explore molecular diversity[J]. Hepatology, 2021, 73(Suppl 1): 14-26. http://www.researchgate.net/publication/348322949_Genetics_of_Hepatocellular_Carcinoma_Approaches_to_Explore_Molecular_Diversity
[8] GEELEHER P, COX N, HUANG R S. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels[J]. PLoS One, 2014, 9(9): e107468. doi: 10.1371/journal.pone.0107468
[9] PAN Y S, CHEN H R, YU J. Biomarkers in hepatocellular carcinoma: current status and future perspectives[J]. Biomedicines, 2020, 8(12): 576. doi: 10.3390/biomedicines8120576
[10] OURA K, MORISHITA A, MASAKI T. Molecular and functional roles of microRNAs in the progression of hepatocellular carcinoma-A review[J]. Int J Mol Sci, 2020, 21(21): 8362. doi: 10.3390/ijms21218362
[11] KIM E, VIATOUR P. Hepatocellular carcinoma: old friends and new tricks[J]. Exp Mol Med, 2020, 52(12): 1898-1907. doi: 10.1038/s12276-020-00527-1
[12] MUINAO T, DEKA BORUAH H P, PAL M. Multi-biomarker panel signature as the key to diagnosis of ovarian cancer[J]. Heliyon, 2019, 5(12): e02826. doi: 10.1016/j.heliyon.2019.e02826
[13] FOUNTZILAS C, KAKLAMANI V G. Multi-gene panel testing in breast cancer management[J]. Cancer Treat Res, 2018, 173: 121-140.
[14] YANG Z C, ZI Q, XU K, et al. Development of a macrophages-related 4-gene signature and nomogram for the overall survival prediction of hepatocellular carcinoma based on WGCNA and LASSO algorithm[J]. Int Immunopharmacol, 2021, 90: 107238. doi: 10.1016/j.intimp.2020.107238
[15] LIU G M, XIE W X, ZHANG C Y, et al. Identification of a four-gene metabolic signature predicting overall survival for hepatocellular carcinoma[J]. J Cell Physiol, 2020, 235(2): 1624-1636. doi: 10.1002/jcp.29081
[16] LIN P, HE R Q, DANG Y W, et al. An autophagy-related gene expression signature for survival prediction in multiple cohorts of hepatocellular carcinoma patients[J]. Oncotarget, 2018, 9(25): 17368-17395. doi: 10.18632/oncotarget.24089
[17] LI G X, XU W Q, ZHANG L, et al. Development and validation of a CIMP-associated prognostic model for hepatocellular carcinoma[J]. EBioMedicine, 2019, 47: 128-141. doi: 10.1016/j.ebiom.2019.08.064
[18] YU J, WU X L, LV M, et al. A model for predicting prognosis in patients with esophageal squamous cell carcinoma based on joint representation learning[J]. Oncol Lett, 2020, 20(6): 387. http://www.ncbi.nlm.nih.gov/pubmed/33193847
[19] YANG Y J, WANG C Y, WEI N D, et al. Identification of prognostic chromatin-remodeling genes in clear cell renal cell carcinoma[J]. Aging (Albany NY), 2020, 12(24): 25614-25642.
[20] HONG W F, LIANG L, GU Y J, et al. Immune-related lncRNA to construct novel signature and predict the immune landscape of human hepatocellular carcinoma[J]. Mol Ther Nucleic Acids, 2020, 22: 937-947. doi: 10.1016/j.omtn.2020.10.002
[21] 陈懿, 李雪, 林文雅, 等. 自噬基因预测肝癌患者长期生存及通路分析[J]. 医学研究杂志, 2021, 50(1): 137-141. https://www.cnki.com.cn/Article/CJFDTOTAL-YXYZ202101031.htm [22] 段万里, 任伟, 邓骞, 等. 基于TCGA数据库的肾癌自噬相关基因预后模型的建立与应用[J]. 现代泌尿外科杂志, 2020, 25(10): 870-875, 889. doi: 10.3969/j.issn.1009-8291.2020.10.003 [23] TABUSE M, OHTA S, OHASHI Y, et al. Functional analysis of HOXD9 in human gliomas and glioma cancer stem cells[J]. Mol Cancer, 2011, 10: 60. doi: 10.1186/1476-4598-10-60
[24] LONG J Y, ZHANG L, WAN X S, et al. A four-gene-based prognostic model predicts overall survival in patients with hepatocellular carcinoma[J]. J Cell Mol Med, 2018, 22(12): 5928-5938. doi: 10.1111/jcmm.13863
[25] LV X P, LI L L, LV L, et al. HOXD9 promotes epithelial-mesenchymal transition and cancer metastasis by ZEB1 regulation in hepatocellular carcinoma[J]. J Exp Clin Cancer Res, 2015, 34: 133. doi: 10.1186/s13046-015-0245-3
[26] CHRISTIANSEN A, DYRSKJOT L. The functional role of the novel biomarker karyopherin α 2 (KPNA2) in cancer[J]. Cancer Lett, 2013, 331(1): 18-23. doi: 10.1016/j.canlet.2012.12.013
[27] GUO X G, WANG Z H, ZHANG J N, et al. Upregulated KPNA2 promotes hepatocellular carcinoma progression and indicates prognostic significance across human cancer types[J]. Acta Biochim Biophys Sin (Shanghai), 2019, 51(3): 285-292. doi: 10.1093/abbs/gmz003
[28] JIANG P, TANG Y Q, HE L, et al. Aberrant expression of nuclear KPNA2 is correlated with early recurrence and poor prognosis in patients with small hepatocellular carcinoma after hepatectomy[J]. Med Oncol, 2014, 31(8): 1-7.
[29] KONIROVA J, OLTOVA J, CORLETT A, et al. Modulated DISP3/PTCHD2 expression influences neural stem cell fate decisions[J]. Sci Rep, 2017, 7: 41597. doi: 10.1038/srep41597
[30] XIE G X, WANG X N, HUANG F J, et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis[J]. Int J Cancer, 2016, 139(8): 1764-1775. doi: 10.1002/ijc.30219
[31] CHEN W B, OU M L, TANG D E, et al. Identification and validation of immune-related gene prognostic signature for hepatocellular carcinoma[J]. J Immunol Res, 2020, 2020: 5494858. http://qikan.cqvip.com/Qikan/Article/Detail?id=7106408960
[32] STACEY D, KAZLAUSKAS A. Regulation of Ras signaling by the cell cycle[J]. Curr Opin Genet Dev, 2002, 12(1): 44-46. doi: 10.1016/S0959-437X(01)00262-3
[33] MATSUDA Y. Molecular mechanism underlying the functional loss of cyclindependent kinase inhibitors p16 and p27 in hepatocellular carcinoma[J]. World J Gastroenterol, 2008, 14(11): 1734-1740. doi: 10.3748/wjg.14.1734
[34] MATSUDA Y, ICHIDA T. p16 and p27 are functionally correlated during the progress of hepatocarcinogenesis[J]. Med Mol Morphol, 2006, 39(4): 169-175. doi: 10.1007/s00795-006-0339-2
[35] GREENBAUM L E. Cell cycle regulation and hepatocarcinogenesis[J]. Cancer Biol Ther, 2004, 3(12): 1200-1207. doi: 10.4161/cbt.3.12.1392
-
期刊类型引用(0)
其他类型引用(1)




计量
- 文章访问数: 342
- HTML全文浏览量: 501
- PDF下载量: 39
- 被引次数: 1
 苏公网安备 32100302010246号
苏公网安备 32100302010246号
